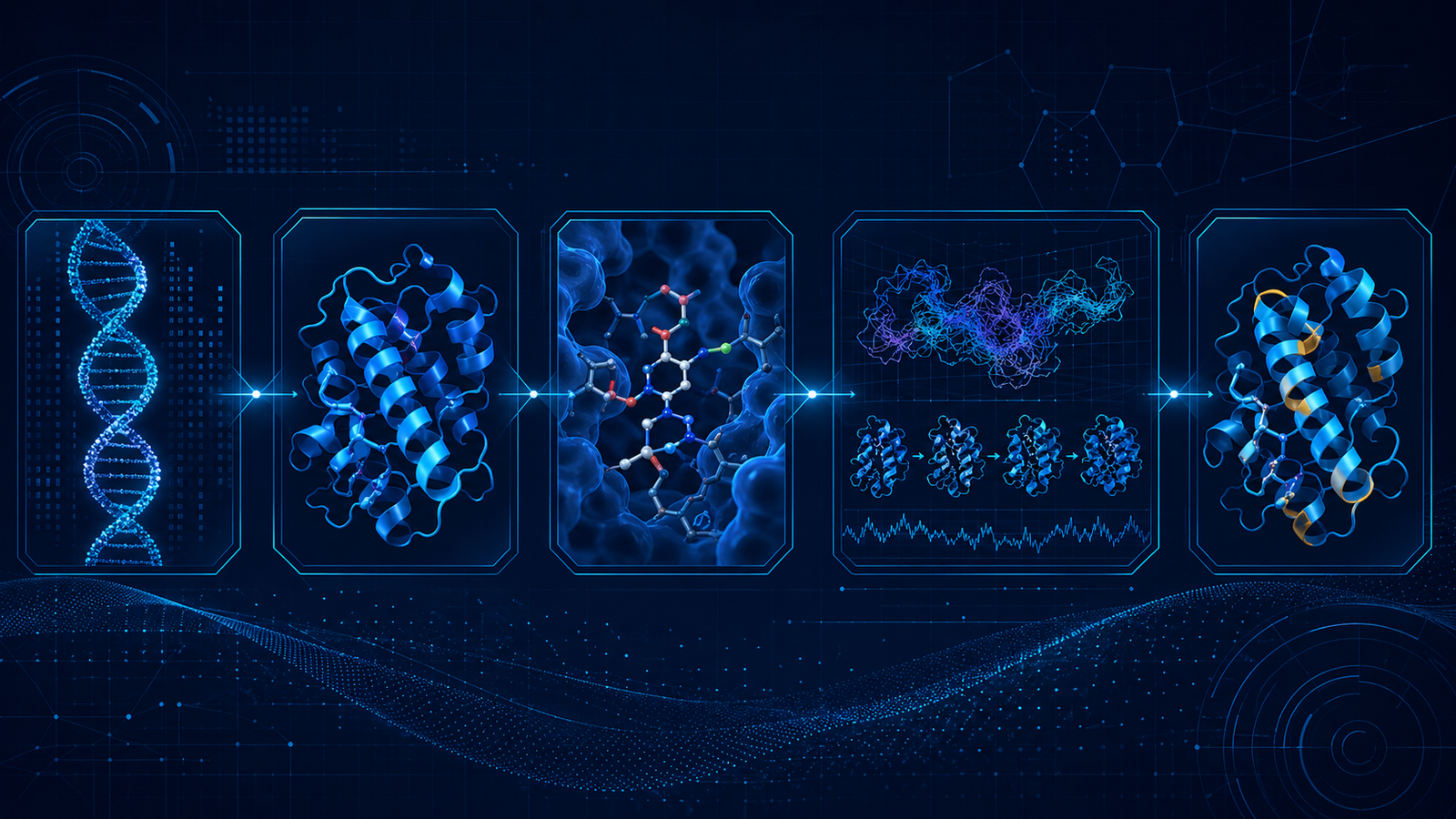
Computational workflows have become central to enzyme discovery and engineering because they allow researchers to evaluate sequence, structural, and mechanistic information before beginning laboratory experiments. Rather than relying only on trial-and-error screening, computational approaches help identify promising enzyme candidates, prioritize mutation sites, predict substrate interactions, and generate testable biological hypotheses (Damborsky & Brezovsky, 2014; Chowdhury & Maranas, 2020; Ferreira et al., 2022). In a typical workflow, the starting point may be an amino-acid sequence, a predicted or experimentally solved structure, a target substrate, or a family of homologous enzymes. The broader goal is to convert digital biological information into experimentally verifiable design decisions.
Sequence analysis is often the first stage of this process. By comparing homologous enzymes through multiple sequence alignment and evolutionary analysis, researchers can identify conserved residues, variable regions, catalytic motifs, and functionally important sequence patterns. Highly conserved residues often indicate structural or catalytic importance, whereas variable regions may reveal positions that tolerate mutation and can therefore be explored for engineering purposes. This information helps guide rational or semi-rational mutagenesis while reducing the risk of disrupting essential catalytic or structural features (Damborsky & Brezovsky, 2014; Monza et al., 2017; Chowdhury & Maranas, 2020).
Structure prediction adds a three-dimensional framework to sequence-based observations. Predicted models can help locate active-site residues, substrate-access channels, flexible loops, domain boundaries, and surface regions that may influence solubility or stability. Recent advances in protein-structure prediction have greatly expanded the availability of structural models for enzymes that lack experimentally determined structures, making structure-guided enzyme engineering more accessible (Jumper et al., 2021; Marques et al., 2021; Ferreira et al., 2022). Although predicted structures should be interpreted with caution, especially for flexible loops, ligand-bound conformations, and multimeric states, they remain valuable for hypothesis generation and experimental planning.
Molecular docking is commonly used to investigate how substrates, inhibitors, or reaction intermediates may bind within an enzyme active site. Docking can suggest likely binding orientations, identify residues involved in substrate recognition, and reveal steric or electrostatic features that may limit catalytic efficiency or substrate scope. However, docking scores are approximate, and many protocols simplify enzyme flexibility, solvent effects, and catalytic geometry. For this reason, docking is most informative when combined with structural knowledge, mechanistic reasoning, and biochemical validation rather than treated as a final prediction (Sirin et al., 2014; Yao et al., 2016; Ferreira et al., 2022).
Molecular dynamics simulations complement static structural models by introducing time-dependent motion. These simulations can reveal how enzymes fluctuate, how active-site residues move, how loops open or close, and how mutations may alter stability, flexibility, or substrate-access pathways. Molecular dynamics is particularly useful for examining conformational states that are not obvious from a single structure, but its conclusions depend on simulation length, force-field quality, sampling depth, and the biological relevance of the modeled system (Osuna et al., 2015; Wang et al., 2023; Zhou & Huang, 2024). Therefore, simulations should be used to identify trends and hypotheses rather than to replace experimental testing.
A robust computational workflow ultimately depends on laboratory validation. Predictions become meaningful only when tested through cloning, protein expression, purification, activity assays, kinetic analysis, thermostability measurements, substrate-scope evaluation, and, where possible, structural characterization. The most effective enzyme-engineering strategies are therefore iterative: computational analysis guides experimental design, experimental data reveal which predictions are correct, and those results inform the next round of modeling and engineering (Honarparvar et al., 2014; Scherer et al., 2021; Nam et al., 2024). In this sense, the future of enzyme engineering lies not in computation alone, but in integrated workflows that combine digital exploration with rigorous biochemical evidence.
References
Chowdhury, R., & Maranas, C. D. (2020). From directed evolution to computational enzyme engineering—A review. AIChE Journal. https://aiche.onlinelibrary.wiley.com/doi/abs/10.1002/aic.16847
Damborsky, J., & Brezovsky, J. (2014). Computational tools for designing and engineering enzymes. Current Opinion in Chemical Biology. https://www.sciencedirect.com/science/article/pii/S1367593113002354
Ferreira, P., Fernandes, P. A., & Ramos, M. J. (2022). Modern computational methods for rational enzyme engineering. Chem Catalysis. cell.com)00528-0
Honarparvar, B., Govender, T., Maguire, G. E. M., Soliman, M. E. S., & Kruger, H. G. (2014). Integrated approach to structure-based enzymatic drug design: Molecular modeling, spectroscopy, and experimental bioactivity. Chemical Reviews. pubs.acs.org
Jumper, J., Evans, R., Pritzel, A., Green, T., Figurnov, M., Ronneberger, O., et al. (2021). Highly accurate protein structure prediction with AlphaFold. Nature. https://doi.org/10.1038/s41586-021-03819-2
Marques, S. M., Planas-Iglesias, J., & Damborsky, J. (2021). Web-based tools for computational enzyme design. Current Opinion in Structural Biology. sciencedirect.com
Monza, E., Acebes, S., Lucas, M. F., & Guallar, V. (2017). Molecular modeling in enzyme design, toward in silico guided directed evolution. In Directed Enzyme Evolution: Advances and Applications. https://link.springer.com/chapter/10.1007/978-3-319-50413-1_10
Nam, K., Shao, Y., Major, D. T., & Wolf-Watz, M. (2024). Perspectives on computational enzyme modeling: From mechanisms to design and drug development. ACS Omega. https://pubs.acs.org/doi/abs/10.1021/acsomega.3c09084
Osuna, S., Jiménez-Osés, G., Noey, E. L., & Houk, K. N. (2015). Molecular dynamics explorations of active site structure in designed and evolved enzymes. Accounts of Chemical Research. https://pubs.acs.org/doi/abs/10.1021/ar500452q
Scherer, M., Fleishman, S. J., & Jones, P. R. (2021). Computational enzyme engineering pipelines for optimized production of renewable chemicals. Frontiers in Bioengineering and Biotechnology. https://www.frontiersin.org/journals/bioengineering-and-biotechnology/articles/10.3389/fbioe.2021.673005/full
Sirin, S., Kumar, R., & Martinez, C. (2014). A computational approach to enzyme design: Predicting ω-aminotransferase catalytic activity using docking and MM-GBSA scoring. Journal of Chemical Information and Modeling. https://pubs.acs.org/doi/abs/10.1021/ci5002185
Wang, P., Zhang, J., Zhang, S., & Lu, D. (2023). Using high-throughput molecular dynamics simulation to enhance the computational design of kemp elimination enzymes. Journal of Chemical Information and Modeling. https://pubs.acs.org/doi/abs/10.1021/acs.jcim.3c00002
Yao, Z., Zhang, L., Gao, B., Cui, D., & Wang, F. (2016). A semiautomated structure-based method to predict substrates of enzymes via molecular docking: A case study with Candida antarctica lipase B. Journal of Chemical Information and Modeling. https://pubs.acs.org/doi/abs/10.1021/acs.jcim.5b00585
Zhou, J., & Huang, M. (2024). Navigating the landscape of enzyme design: From molecular simulations to machine learning. Chemical Society Reviews. https://pubs.rsc.org/en/content/articlehtml/2023/78/d4cs00196f